ATAC-seq测序概述
测序简介
ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) 是一种确定整个基因组染色质可及性的方法。它利用高度活跃的 Tn5 转座酶将测序接头插入开放染色质区域,其原理如下:
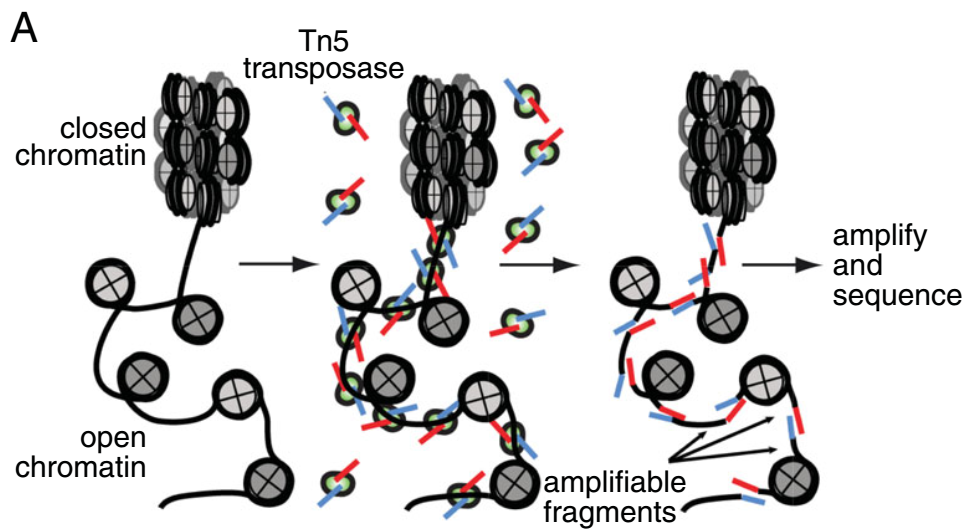
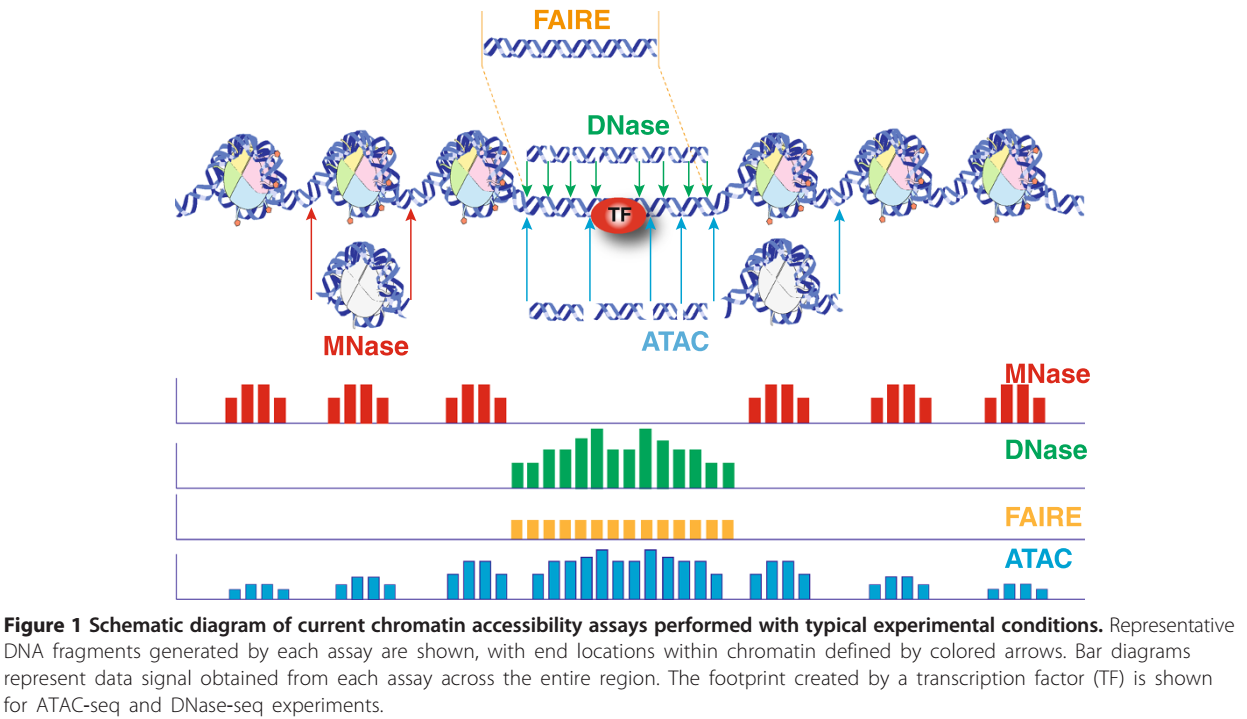
其与其他几种类似的测序手段生成的数据峰图模式如下:
使用途径
- 构建物种表观基因组图谱
- 识别鉴定核小体(nucleosome)的位置
- 识别重要的转录因子
- 构建转录因子结合的footprinting
实验设计
- two or more biological replicates
- each replicate has 25 million non-duplicate, non-mitochondrial aligned reads for single-end sequencing and 50 million for paired-ended sequencing
- typically, no need for background 'input'
- use as few PCR cycles as possible when constructing the library
- paired-end sequencing is preferred
参考
数据处理
Quality control
ATAC-seq同样是二代测序数据,因此可以同样使用Trimmomatic和Trim Galore进行质控和去接头
比对和过滤
比对
# better alignment results are frequently achieved with --very-sensitive
# use -X 2000 to allow larger fragment size (default is 500)
bowtie2 --very-sensitive -X 2000 -x $Bowtie2Index -1 ${sample}*_1.fq.gz -2 ${sample}*_2.fq.gz -p 10 2> $${sample}.bowtie2.log |
samtools sort -@ 10 -O bam -o ${sample}.sorted.bam
samtools index -@ 10 $WORKDIR/bowtie2/${sample}.sorted.bam线粒体read过滤
由于线粒体基因组中没有感兴趣的 ATAC-seq 峰,这些读数只会使后续步骤复杂化。因此,我们建议通过以下方法之一将它们从进一步分析中删除:
- 在进行比对之前建议将参考基因组中的线粒体基因组删除。一般在基因组中线粒体染色体被标记为chrM,如果在构建基因组索引之前从参考序列中删除,可能会时比对率降低
- 比对之后删除线粒体read,可以通过samtools等工具进行删除
samtools view -@ 10 -h ${sample}.bam | grep -v chrM | samtools sort -@ 10 -O bam -o ${sample}.rmChrM.bam去除PCR重复
PCR dupliate 是 PCR 过程中产生的 DNA 片段的copy。由于它们是测序文库制备过程的产物,所以可能会干扰感兴趣的生物信号。因此需要被删除
可以使用 picard的 MarkDuplicates标记PCR dupliate,然后使用 samtools删除
# REMOVE_DUPLICATES=false: mark duplicate reads, not remove.
# Change it to true to remove duplicate reads.
java -XX:ParallelGCThreads=10 -Djava.io.tmpdir=/tmp -jar picard MarkDuplicates \
QUIET=true INPUT=${sample}.bam OUTPUT=${sample}.marked.bam METRICS_FILE=${sample}.sorted.metrics \
REMOVE_DUPLICATES=false CREATE_INDEX=true VALIDATION_STRINGENCY=LENIENT TMP_DIR=/tmp
## dupliate reads labeled as 1024
samtools view -h -b -F 1024 ${sample}.bam > ${sample}.rmDup.bamNon-unique alignments过滤
MAPQ值可以衡量read的比对结果,通过该值可以过滤比对质量差或者说非unique比对的read
注意
不同的基因组比对软件实现MAPQ的方式可能不同,因此需要注意在不同软件中的阈值,一般情况下选用MAPQ > 30作为阈值
# Remove multi-mapped reads (i.e. those with MAPQ < 30, using -q in SAMtools)
samtools view -h -q 30 ${sample}.bam > ${sample}.rmMulti.bam注意
关于比对read的过滤,有一些会进行更加严格的过滤,只保留properly mapped reads,可以借助Decoding SAM flags进行flag的选取
合并bam文件
当针对一种实验条件重复构建多个文库时,可以在calling peak之前合并不同样本的BAM文件(例如,合并技术重复中的 BAM 文件、合并 BAM 文件会获得更大的测序深度)
合并 bam文件可以使用 samtools
samtools merge -@ 10 condition1.merged.bam sample1.bam sample2.bam sample3.bam
samtools index -@ 10 condition1.merged.bampeak calling
对于peak calling,一般情况下我们使用MACS进行分析。MACS最初开发是针对CHIP-seq进行使用的,因此有些参数对于ATAC-seq数据来说并不一定合适。一般情况下需要设置 --nomodel参数,因为 MACS 的shifting model对于开放染色数据实际上没有意义。由于一般是双末端测序数据,所以可以设置 -f BAMPE,它会让MACS考虑真实的read片段长度而不是通过计算估计的长度。
# -f BAMPE, use paired-end information
# --keep-dup all, keep all duplicate reads.
macs2 callpeak -f BAMPE -g hs --keep-dup all --cutoff-analysis -n sample1 -t sample1.shifted.bam --outdir macs2/sample1 2> macs2.log创建bigwig文件
bigwig文件可以用于后续的IGV可视化等,如果使用 macs calling peak,可以直接添加 -B参数生成bedGraph文件,之后使用bedGraphToBigWig等工具进行转换。
同样的 deeptools是一个用于表观基因组可视化的工具,可以使用其中的 bamCoverage创建bigwig文件,它提供了几种不同的方式来对read进行标准化
# bam to bigwig, normalize using 1x effective genome size
# effective genome size: https://deeptools.readthedocs.io/en/latest/content/feature/effectiveGenomeSize.html
bamCoverage --numberOfProcessors 8 --binSize 10 --normalizeUsing RPGC --effectiveGenomeSize $effect_genome_size --bam sample1.shifted.bam -o sample1.shifted.bw质量检查
在进行 ATAC-seq 数据的分析时,我们需要使用一些指标检查数据或者是构建文库的质量,以便对数据的质量进行控制。
ENCODE计划进行了大量的ATAC-seq数据的检测,给出了一套比较规范的ATAC-seq实验的标准和指控指标。

Fragment/Insert size
一种常见的数据质量控制是绘制构建的文库中片段的大小密度。 ATAC 文库的成功构建需要 DNA 末端有一对适当的 Tn5 转座酶切割事件。在无核小体的开放染色质区域,许多 Tn5 分子可以介入并将 DNA 切成小片段;在核小体占据区域周围,Tn5 只能访问连接子区域。因此,在正常的 ATAC-seq 文库中,您应该会在 < 100 bp 的区域(开放染色质)看到尖峰,在 ~200bp 区域(单核小体)看到尖峰,以及其他较大的峰(多核小体),如下图所示:
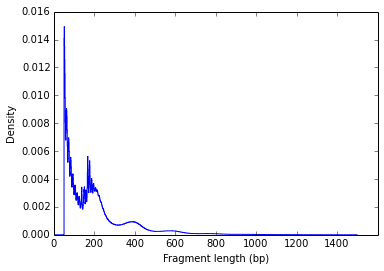
这种清晰的核小体定相模式表明实验质量良好。
该图对应的数据可以使用以下代码来计算
samtools view ATAC_sorted.bam | awk '$9>0' | cut -f 9 | sort | uniq -c | sort -b -k2,2n | sed -e 's/^[ \t]*//' > fragment_length_count.txt参考:
Biostars - ATAC-seq fragment length distributionNot A Rocket Scientist - ATAC-seq insert size plotting
mitochondrial reads
正常测序情况下,不能出现太高的线粒体read比例,否则会干扰到后续的分析,可以使用以下方式计算线粒体read的比例
samtools index ATAC_sorted.bam
mtReads=$(samtools idxstats ATAC_sorted.bam | grep 'chrM' | cut -f 3)
totalReads=$(samtools idxstats ATAC_sorted.bam | awk '{SUM += $3} END {print SUM}')
echo '==> mtDNA Content:' $(bc <<< "scale=2;100*$mtReads/$totalReads")'%'Library complexity
文库复杂度表示的大致原理是检测到的unique的read占整体或者是特定read的比例,反映的是文库的覆盖程度。可以使用下边的Non-Redundant Fraction (NRF) and PCR Bottlenecking Coefficients 1 and 2来计算
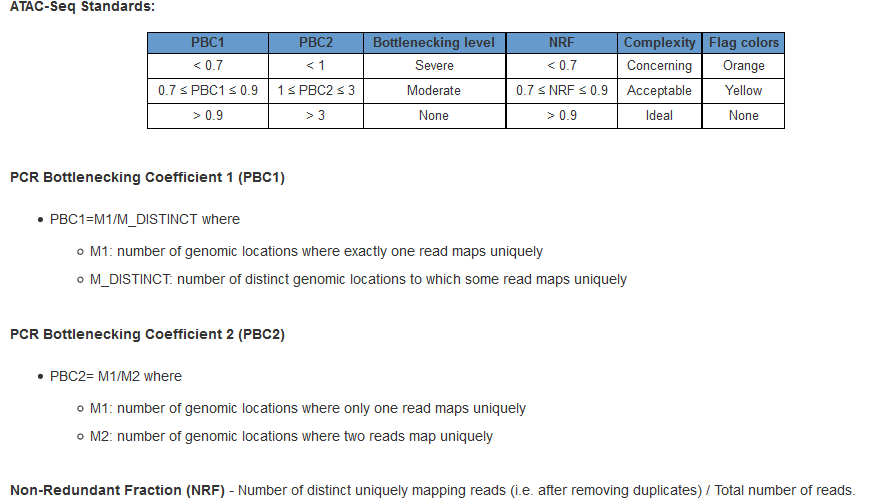
# the bam used here is sorted bam after duplicates marking
# sort bam by names
samtools sort -@ 10 -n -O BAM -o tmp.bam ATAC.bam
# calculate PBC metrics
bedtools bamtobed -bedpe -i tmp.bam | awk 'BEGIN{OFS="\t"}{print $1,$2,$4,$6,$9,$10}' \
| grep -v 'chrM' | sort | uniq -c | awk 'BEGIN{mt=0;m0=0;m1=0;m2=0}($1==1){m1=m1+1} \
($1==2){m2=m2+1} {m0=m0+1} {mt=mt+$1} \
END{printf "%d\t%d\t%d\t%d\t%f\t%f\t%f\n", mt,m0,m1,m2,m0/mt,m1/m0,m1/m2}' > ATAC_sorted.pbc.qc
rm tmp.bamFRiP score
FRiP score全称Fraction of reads in peaks,是指落入所谓峰区域的所有映射read的分数,即显着富集峰中的可用read除以所有可用read。FRiP score理论上应该大于0.3(在一定程度上表示的是富集的水平),但大于0.2的值是可以接受的。
FRiP score可以通过 samtools和 bedtools计算:
# total reads
total_reads=$(samtools view -c ATAC.bam)
# reads in peaks
reads_in_peaks=$(bedtools sort -i ATAC_peaks.narrowPeak \
| bedtools merge -i stdin | bedtools intersect -u -nonamecheck \
-a ATAC.bam -b stdin -ubam | samtools view -c)
# FRiP score
FRiP=$(awk "BEGIN {print "${reads_in_peaks}"/"${total_reads}"}")Blacklist
Blacklist是指人类基因组中的一组特定区域,该区域在二代测序实验中往往具有异常的,非结构化的高信号或者读数,这些区域与一些染色体原件如微卫星,着丝粒和端粒相重合。ENCODE计划鉴定识别了这些区域,可以在分析过程中将其过滤掉。
merge peaks
一般情况下,一组样本可能有2-3个重复。在分析时,我们希望把相同组或者条件下的峰进行合并。下边是Corces et al., 2016文章的做法。
To generate a non-redundant list of hematopoiesis- and cancer-related peaks, we first extended summits to 500-bp windows (±250 bp). We then ranked the 500-bp peaks by summit significance value (defined by MACS2) and chose a list of non-overlapping, maximally significant peaks.
具体代码可以用以下方式实现:
## reference: https://bedops.readthedocs.io/en/latest/content/usage-examples/master-list.html
summit_bed=(sample1_summits.bed sample2_summits.bed sample3_summits.bed)
out=fAdrenal.master.merge.bed
tmpd=/tmp/tmp$$
mkdir -p $tmpd
## First, union all the peaks together into a single file.
bedlist=""
for bed in ${beds[*]}
do
bedlist="$bedlist $summit_bed"
done
# extended summits to 500-bp windows (±250 bp)
bedops --range 250 -u $bedlist > $tmpd/tmp.bed
## The master list is constructed iteratively. For each pass through
## the loop, elements not yet in the master list are merged into
## non-overlapping intervals that span the union (this is just bedops
## -m). Then for each merged interval, an original element of highest
## score within the interval is selected to go in the master list.
## Anything that overlaps the selected element is thrown out, and the
## process then repeats.
iters=1
solns=""
stop=0
while [ $stop == 0 ]
do
echo "merge steps..."
## Condense the union into merged intervals. This klugey bit
## before and after the merging is because we don't want to merge
## regions that are simply adjacent but not overlapping
bedops -m --range 0:-1 $tmpd/tmp.bed \
| bedops -u --range 0:1 - \
> $tmpd/tmpm.bed
## Grab the element with the highest score among all elements forming each interval.
## If multiple elements tie for the highest score, just grab one of them.
## Result is the current master list. Probably don't need to sort, but do it anyway
## to be safe since we're not using --echo with bedmap call.
bedmap --max-element $tmpd/tmpm.bed $tmpd/tmp.bed \
| sort-bed - \
> $tmpd/$iters.bed
solns="$solns $tmpd/$iters.bed"
echo "Adding `awk 'END { print NR }' $tmpd/$iters.bed` elements"
## Are there any elements that don't overlap the current master
## list? If so, add those in, and repeat. If not, we're done.
bedops -n 1 $tmpd/tmp.bed $tmpd/$iters.bed \
> $tmpd/tmp2.bed
mv $tmpd/tmp2.bed $tmpd/tmp.bed
if [ ! -s $tmpd/tmp.bed ]
then
stop=1
fi
((iters++))
done
## final solution
bedops -u $solns \
> $out
## Clean up
rm -r $tmpd
exit 0当然也可以根据peak的overlap情况直接进行合并