Skip to content 
此页内容
测序深度和覆盖度
测序深度 && 覆盖度
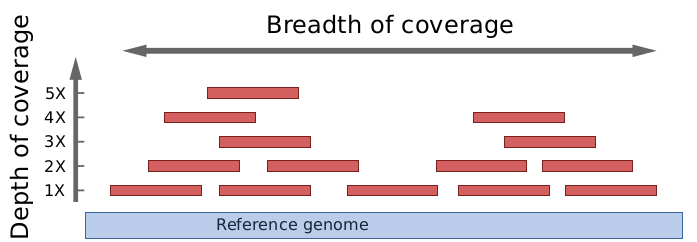
测序深度 (Depth)
基因组被测序片段(短读 short reads)“覆盖”的强度有多大?
每一碱基的覆盖率是基因组碱基被测序的平均次数。 基因组的覆盖深度是通过与基因组匹配的所有短读的碱基数目除以该基因组的长度来计算的。 它通常表示为1X、2X、3X、…(1、2或3倍覆盖)。
这里的覆盖率通常被称作测序深度(sequencing depth)或者覆盖深度(depth of coverage)
测序深度的计算公式如下:
- 原始测序数据的深度: Depth = (# sequenced bases) / (# bases of reference)
- 比对上的数据的深度: Depth= (# bases of all mapped reads) / (# bases of reference)
覆盖度 (coverage)
短读“覆盖”了基因组的多少区域?是否有些区域没有任何的短读覆盖?
覆盖范围是参考基因组被一定深度覆盖的碱基的百分比。例如,一个基因组的90%区域在1X深度覆盖,另外仍然有70%的区域被覆盖了5X深度。
此处通常称为覆盖度(genome covarage)或者覆盖范围(breadth of coverage)。
覆盖度的计算公式如下:
Coverage = (# area covered by mapped reads) / (# area of reference)
计算示例
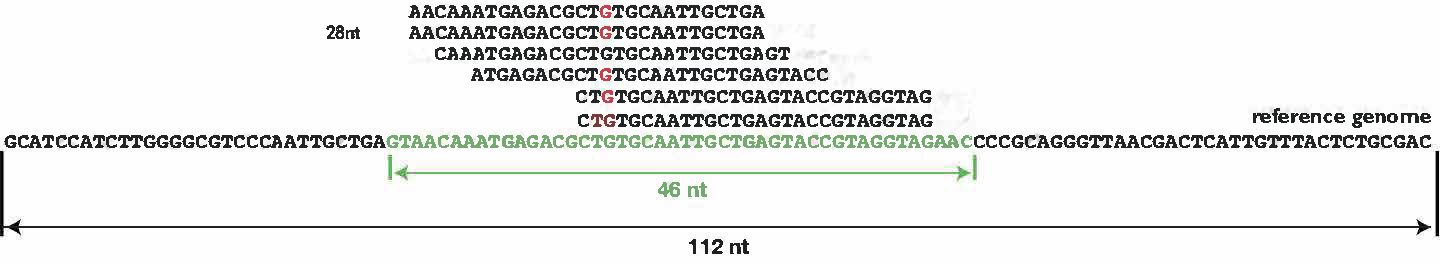
对于全基因组范围:
Depth = (6 * 28nt) / 112nt = 1.2 fold
Coverage = (46nt - 5nt) / 112nt = 36.6%
对于target区域:
Depth = (6 * 28nt) / 46nt = 3.7 fold
Coverage = (46nt - 5nt) / 46nt = 89.1%
计算测序深度和覆盖度
samtools
## 计算每个位点的测序深度
samtools depth samp.bam > base_depth.txt
## 统计平均覆盖度
cat base_depth.txt | awk '{ sum += $3; } END { print "coverage = " sum/NR }'BAMStats
sambamba sort -o smap.sort.bam -t 20 -p samp.bam
java -jar -Xmx30g BAMStats-1.25.jar -i samp.sort.bam -o BAMStats --view simple
cat BAMStats| ref | N | mean | median | sd | q1 | q3 | 2.50% | 97.50% | min | max |
|---|---|---|---|---|---|---|---|---|---|---|
| chr1 | 249,250,621 | 0.63 | 0 | 11.29 | 0 | 1 | 0 | 3 | 0 | 15051 |
| chr2 | 243,199,373 | 0.63 | 0 | 1.43 | 0 | 1 | 0 | 3 | 0 | 706 |
| chr3 | 198,022,430 | 0.64 | 0 | 1 | 0 | 1 | 0 | 3 | 0 | 102 |
| chr4 | 191,154,276 | 0.58 | 0 | 1.13 | 0 | 1 | 0 | 3 | 0 | 527 |
Qualimap
qualimap bamqc --java-mem-size=40G -c -nt 30 -outdir ./ -outformat PDF -os -outfile samp.bamqc.pdf -bam samp.sort.bam -gd hg19 -nr 2000