eggnog-mapper功能注释
eggNOG-Mapper介绍
通常功能注释的思路都是基于序列相似性找直系同源基因,常见的方法就是BLAST+BLAST2GO, 或者是InterProScan。eggNOG-mapper的作者认为这种方法不够可靠,毕竟你有可能找到的的是旁系同源基因。近期对多个工具的整体评估发现eggNOG(evolutionary genealogy of genes: Non-supervised Orthologous Groups)在区分旁系同源基因和直系同源基因上表现不错,因此基于eggNOG数据库开发了eggNOG-mapper工具,用于对新序列进行功能注释。
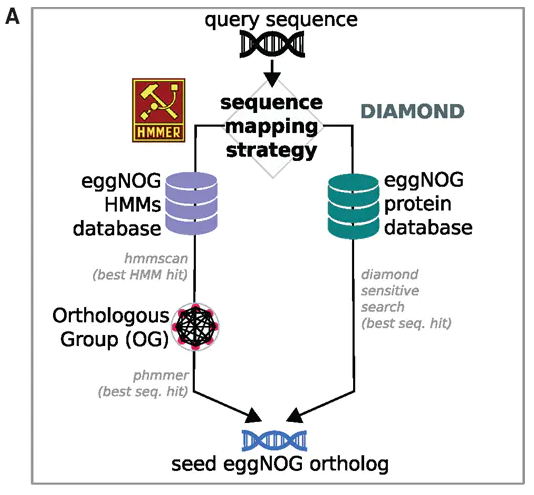
eggNOG-mapper的算法实现如下:
- 序列比对。首先,每条蛋白序列用HMMER3在整理的eggNOG数据库中搜索。由于每个HMM匹配都和一个功能注释的eggNOG OG对应,这一步就提供了初步的注释信息,
- 每条蛋白序列用phmmer在最佳匹配的HMM对应的一组eggNOG蛋白中进一步搜索,
- 每条序列的最佳匹配结果以 seed ortholog 形式存放,用于获取其他直系同源基因。
目前eggNOG HMM数据库中拥有1,911,745个OG,覆盖了1,678种细菌,115种古细菌,238种真核物种以及352种病毒。除了HMMER3外,还而可用DIAMOND直接对所有的eggNOG蛋白序列进行搜索,它的速度更快,适合类似于宏基因组这类大数据集,或者是已有物种和eggNOG所收集的物种比较近
- 推测直系同源基因。每个用于检索的蛋白序列的最佳匹配序列会对应eggNOG的一个蛋白, 这些蛋白基于预分析的eggNOG进化树数据库会提取一组更加精细的直系同源基因。这一步还会根据bit-screo或E-value对结果进行一次过来,剔除同源性不高的结果
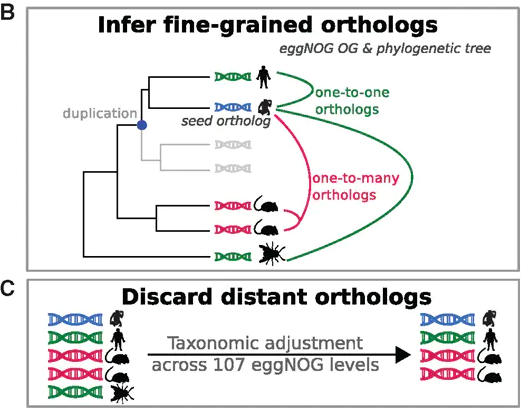
- 功能注释。用于搜索的蛋白序列对应的直系同源基因的功能描述就是最终的注释结果。比如说GO, KEGG, COG等。

安装
安装本体eggnog-mapper之前,需要先保证服务器上已经安装Python3.7(BioPython模块), wget, HMMER3 和/或 DIAMON,此外你还得保证70G的空间用于存放注释数据库和FASTA文件,对于真核生物至少保证90G的服务器内存。
git clone https://github.com/jhcepas/eggnog-mapper.git之后需要下载所需要的数据库。eggNOG提供了107个不同物种的HMM数据库(xxxNOG)以及三个优化数据库, euk对应真核生物,bact对应细菌, arch对应古细菌(Archeabacteria), 以及一个病毒数据库(viruses). 这三个优化数据库包含了属于该分类内的所有物种的HMM模型。
cd eggnog-mapper
python3 ./download_eggnog_data.py用法
eggnog-mapper用起来非常的简单,你需要提供蛋白序列或基因组序列等作为输入
python3 path/to/emapper.py -i test.fasta --itype genome --cpu 10 -o rapaeggnog-mapper默认是以HMMER进行序列搜索,可以通过-m diamond更改成DIAMOND,参数一般使用默认就好。
--itype表示输入的序列文件类型,可选的有:CDS, proteins, genome, metagenome
--cpu选取线程数目
-o表示输出文件的前缀,默认输出在当前文件夹下
结果
eggnog-mapper会生成三个文件:
- [project_name].emapper.hmm_hits: 记录每个用于搜索序列对应的所有的显著性的eggNOG Orthologous Groups(OG). 所有标记为"-"则表明该序列未找到可能的OG
- [project_name].emapper.seed_orthologs: 记录每个用于搜索序列对的的最佳的OG, 也就是[project_name].emapper.hmm_hits里选择得分最高的结果。之后会从eggNOG中提取更精细的直系同源关系(orthology relationships)
- [project_name].emapper.annotations: 该文件提供了最终的注释结果。大部分需要的内容都可以通过写脚本从从提取,一共有13列。
[project_name].emapper.annotations每一列对应的记录如下:
- query_name: 检索的基因名或者其他ID
- seed_ortholog: eggNOG中最佳的蛋白匹配
- evalue: 最佳匹配的e-value
- score: 最佳匹配的bit-score
- eggNOG_OGs: 预测的基因名,特别指的是类似AP2有一定含义的基因名,而不是AT2G17950这类编号