基础背景

这是一个经典的单细胞分析流程,主要分为以下几个步骤:
实验设计
在正式分析之前,关于实验问题的探讨是很有必要的,最重要的一个就是技术的选择:
- Droplet-based: 10X Genomics, inDrop, Drop-seq
- Plate-based with unique molecular identifiers (UMIs): CEL-seq, MARS-seq
- Plate-based with reads: Smart-seq2
- Other: sci-RNA-seq, Seq-Well
每种方法都有优劣(Mereu et al. 2019; Ziegenhain et al. 2017),目前以10X为代表的droplet-based方法由于高通量和低细胞成本成为了约定俗成的技术;Plate-based方法可以捕获其他的一些表型信息(如细胞形态),另外可以根据实验目的进行调整,灵活性比较好;Read-based方法可以覆盖全转录本,在分析可变剪切、外显子突变等方面很有用;UMI-based方法可以减轻PCR扩增偏差。
下一个问题就是:到底要捕获多少细胞?测序要测多深?
答案很清晰: As much as you can afford to spend.
再补充一下这个答案就是:想要发现罕见细胞类群,就要多获得细胞;想要探索潜在的微小差异,就要加大测序深度。目前常用的droplet-based仪器可以捕获1万到10万细胞,测序深度是每个细胞1000到10000 UMIs,在经济条件一定的前提下,它们之间一般是成反比。另外它还要权衡高细胞捕获通量和影响捕获效率的“双细胞比例”。
实验设计和常规转录组类似,也是要考虑一个实验条件下多个生物重复,而且实验条件最好不要混杂批次。需要注意的是:生物重复不是指的单个细胞,而是指的提供细胞的供体(donors)或者细胞培养体系(cultures)
获得表达矩阵
和常规转录组一样,单细胞转录组也是需要得到表达矩阵,才能进行下游分析。表达矩阵包含的信息就是:每个细胞中比对到每个基因的UMIs或者reads数。有一点需要注意:它的定量方法和具体的实验技术相关
- 10X的数据:使用
CellRanger软件,基于STAR比对到参考基因组,然后统计每个基因的UMIs数量 - Pseudo-alignment方法(如
alevin):不需要比对参考基因组,节省时间、内存 - 对于一些高度multiplexed的方法:可以使用
scPipe包:提供了一套综合的分析流程,利用Rsubread比对,然后统计每个基因的UMIs数量 - CEL-seq、CEL-seq2数据:
scruff包可以专门分析 - read-based方法:可以使用常规bulk 转录组定量的流程(比如smartseq2就可以用hisat2+featureCounts)
- 任何包含spike-in转录本的数据:spike-in序列都要在比对、定量之前加到参考基因组中
multiplexed:翻译叫做”多路复用“,即:large numbers of libraries to be pooled and sequenced simultaneously during a single run,可以节省成本和时间
定量结束后,一般是先导入表达矩阵然后创建一个 SingleCellExperiment对象(例如:read.table() + SingleCellExperiment())。除此以外,还有一些特定的文件格式需要用特定的包,比如DropletUtils可以分析10X数据,tximport/tximeta 可以分析pseudo-alignment数据
需要注意的点:
- 一些定量工具会统计表达矩阵中的reads比对率,会存在一些未必对的情况。尽管这些信息可以用作质控,但这些数值如果被误认为是表达量信息,那么就会干扰下游分析。因此在进行下游分析之前,这部分信息可以去掉或者保存在
colData中 - 如果分析的是人类数据并且加入了ERCC,我们很多时候直接用
^ERCC在行名中进行正则匹配,但是这时要小心,因为ERCC基因家族在人类基因组注释中确实存在,很有可能将真的基因作为外源转录本进行分析。这个问题可以通过将表达矩阵的行名设置为Ensembl,或Entrez来解决
数据处理与下游分析
- quality control:去掉低质量细胞。这些细胞可能在建库环节被破坏,可能没有被有效捕获(这就是所谓的“dropout”)。一般会统计:每个细胞的全部count数、spike-in或线粒体reads比例、检测到基因的数量
- normalized expression value:为了减小细胞文库的偏差(可能由于细胞捕获效率不同、测序深度的差异而造成文库大小差异),把细胞们放在同一起跑线上,才能进行下面的细胞相似性比较,后面再根据相似性进行细胞分群。一般是基于log转换(当然有的函数也涉及了一些size factor的计算),从而对均值-方差进行校正
- feature selection:一般选取高变异基因(Highly Variable Genes)进行后续分析,使用HVGs不用全部基因的原因一是为了减少计算量,二是减少不感兴趣基因(比如在细胞之间没什么差异)对分析产生的噪音
- dimensionality reduction:让数据更“紧凑”,一般是线性降维PCA+非线性降维tSNE/umap。PCA一般是先获得初步的低维数据(可能会挑出几十个主成分),然后传给t-SNE进一步压缩,进行可视化
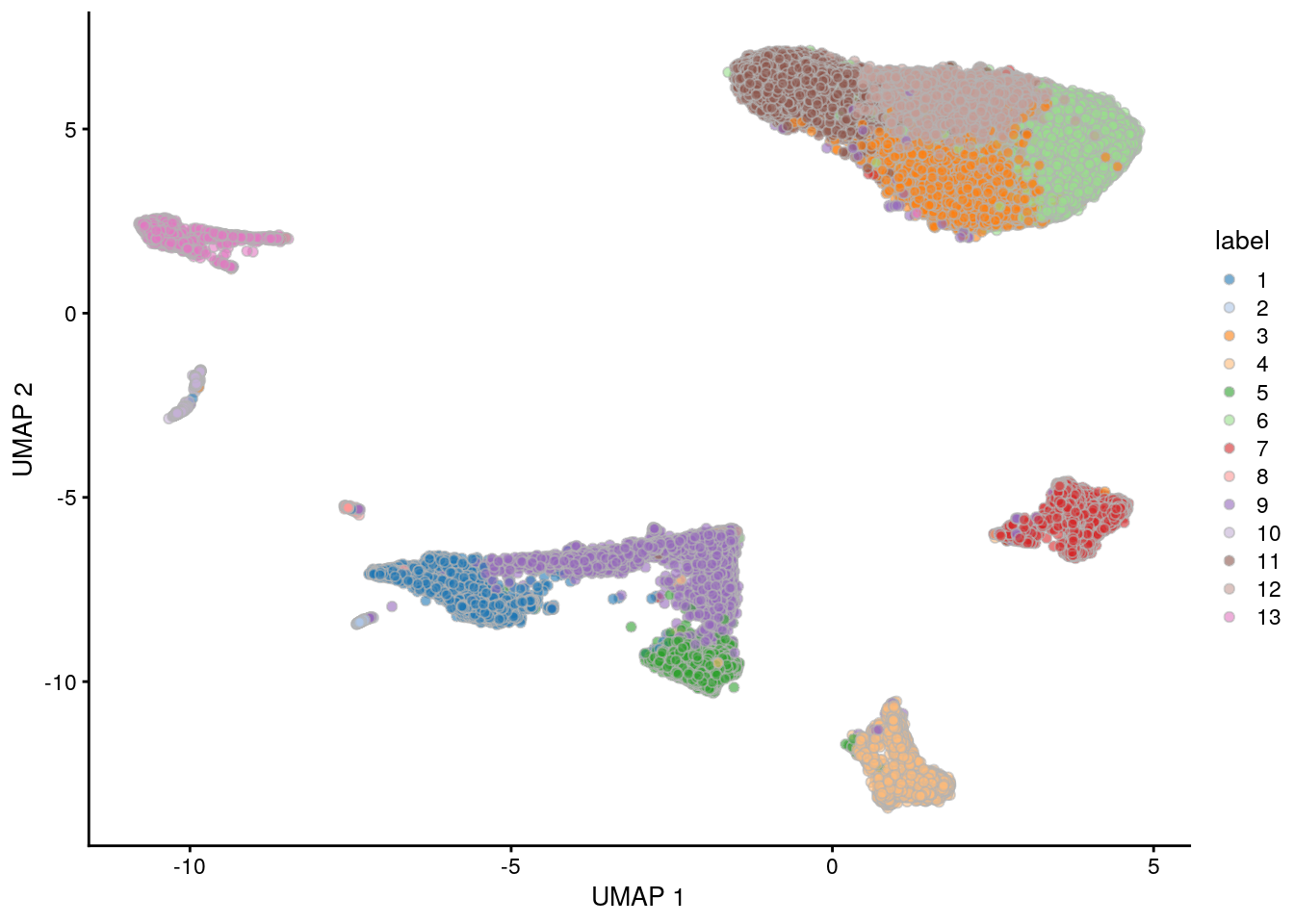
- clustering:根据细胞归一化后的表达量相似性分成组,然后根据每个组marker基因(可理解为这一群细胞的标志性基因)的差异表达对分群进行生物学定义
演示
用 scRNAseq包的droplet-based的视网膜数据【Macosko et al. (2015)】进行演示。
library(scRNAseq)
sce <- MacoskoRetinaData()
# Quality control.
library(scater)
is.mito <- grepl("^MT-", rownames(sce))
qcstats <- perCellQCMetrics(sce, subsets=list(Mito=is.mito))
filtered <- quickPerCellQC(qcstats, percent_subsets="subsets_Mito_percent")
sce <- sce[, !filtered$discard]
# Normalization.
sce <- logNormCounts(sce)
# Feature selection.
library(scran)
dec <- modelGeneVar(sce)
hvg <- getTopHVGs(dec, prop=0.1)
# Dimensionality reduction.
set.seed(1234)
sce <- runPCA(sce, ncomponents=25, subset_row=hvg)
sce <- runUMAP(sce, dimred = 'PCA', external_neighbors=TRUE)
# Clustering.
g <- buildSNNGraph(sce, use.dimred = 'PCA')
colLabels(sce) <- factor(igraph::cluster_louvain(g)$membership)
# Visualization.
plotUMAP(sce, colour_by="label")