Cell Ranger使用基础
背景
Cell Ranger是由10x Genomics公司提供的一套用于处理单细胞RNA测序数据的工具。官网对其进行了详细的介绍和使用说明。可以通过10x Genomics Cloud Analysis使用这个软件或者将其安装到本地。
10X genomics 测序原理
10X简单原理
一个油滴(GEM) = 一个单细胞 + 一个凝胶微珠 = 一次scRNA-seq 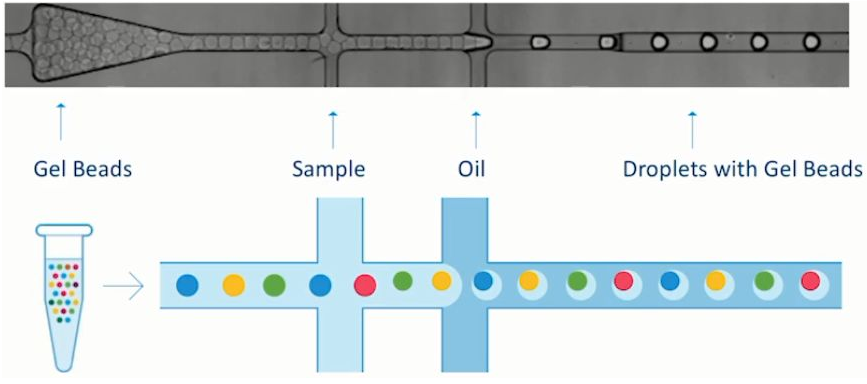
一个单细胞文库包含的微观信息如下: 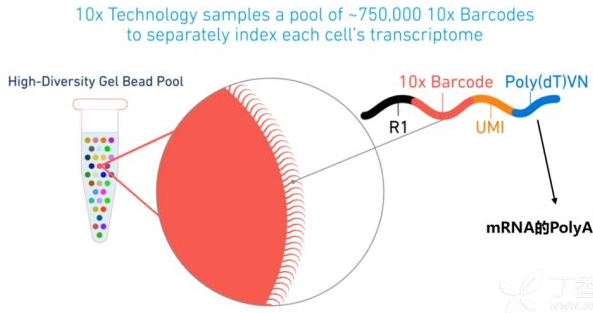
对于V2试剂盒构建的序列结构如下: 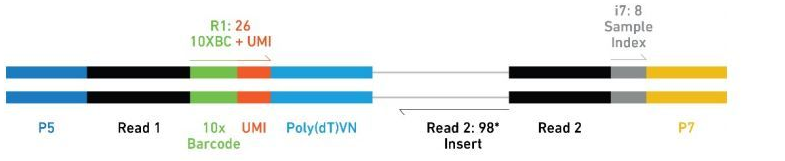
对于V3试剂盒构建的序列结构如下: 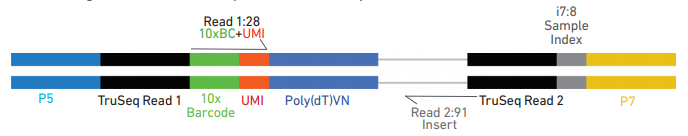
- Barcode: 用来标记细胞
- UMI (Unique Molecular Identifier): 用来标记转录本
- PolyT: 用来捕获成熟的RNA
相关术语
- sample: (样本)从单一来源(比如血液、组织等)提取的细胞悬液
- Library: (文库)单个样本制备的10X barcode 测序文库,对应10X Chromium Controller一个run(即运行一次)的单个芯片通道
- Sequencing Run / Flowcell: 测序仪主要依靠flowcell(例如Hiseq4000就有2个flowcell),每运行一次就是run一次,数据会在flowcell上产出,然后这些数据又会根据flowcell上的不同lane以及lane上不同样本的index进行区分
提示
- 单独一个样本可以制备成多个文库,这样可以一次run就得到更多的细胞数量,而不用在单次run的单个文库中使用全部的样品
- 单独一个样本可以跨多个flowcell测序,如果它们是在一次测序过程中产出,就可以将产出的reads合并
- 一个文库可以用多个flowcell,那么一个flowcell也可以包含多个文库,使用不同的lane上或者样本的index进行区分
- 同一个10x 芯片的两个channels可以说是两个文库,但是两个不同芯片的同一个channel不能说是两个文库
cell ranger 安装
CellRanger必须在Linux系统下运行。首先去下载Linux版本对应的软件安装包,下载之前需要提交信息登陆10x Genomics。然后登录自己的服务器并使用mkdir 和cd等命令建立并进入到软件安装目录。使用下面的命令下载软件:
## 使用 curl
curl -o cellranger-8.0.1.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.1.tar.gz?Expires=1728179900&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=CJ5UER4VwoTgNMsuTsI9Bvv7Y5z0mjcGSDWIxun93bB-NlBZxr8C-vLaUyNfotsFTS3coAllmkcjnhW8rb9EAmA1oLFrNzaoPNmtff3DM1qk02cpdPbiChwXRW6c4NZ1Aw6YEiMoI4-KaYCCz6vrNhZ1NeKoFrM8RthKS5Br8-izW3i3RLdJsxyXW8BjOzKWtWH7rB7J2Mhg~MKez9-XJqMsmYDpSTBam1zFq42x5JMNrcMIuzZAzxunCFFZOPTlLO9RetLNPi2TcldZn2jsl8yuhQPXHqDhyBhysbDIAYodP8EyylYt0lNkuGuigVpqo0e1yyl1aqFG~9VnkExIcQ__"
## 使用 wget 或axel 等
wget -O cellranger-8.0.1.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.1.tar.gz?Expires=1728179900&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=CJ5UER4VwoTgNMsuTsI9Bvv7Y5z0mjcGSDWIxun93bB-NlBZxr8C-vLaUyNfotsFTS3coAllmkcjnhW8rb9EAmA1oLFrNzaoPNmtff3DM1qk02cpdPbiChwXRW6c4NZ1Aw6YEiMoI4-KaYCCz6vrNhZ1NeKoFrM8RthKS5Br8-izW3i3RLdJsxyXW8BjOzKWtWH7rB7J2Mhg~MKez9-XJqMsmYDpSTBam1zFq42x5JMNrcMIuzZAzxunCFFZOPTlLO9RetLNPi2TcldZn2jsl8yuhQPXHqDhyBhysbDIAYodP8EyylYt0lNkuGuigVpqo0e1yyl1aqFG~9VnkExIcQ__"下载完成后解压文件:
tar -xzvf cellranger-x.y.z.tar.gz将cellrange文件添加到环境变量中,即可正常使用cellranger
export PATH="/path/to/cellrange:$PATH"
cellranger -V
# cellranger cellranger-7.2.0使用命令
cellranger提供了五个分析模块用于分析单细胞转录组数据
cellranger mkfastq
该命令封装了illumina的bcl2fastq软件,用来拆分illumina测序仪得到的BCL(raw base call)格式文件,输出fastq文件。
使用方法
## 两种使用方法
cellranger mkfastq --id=tiny-bcl \
--run=/path/to/tiny_bcl \
--csv=cellranger-tiny-bcl-simple-1.2.0.csv
cellranger mkfastq --id=tiny-bcl \
--run=/path/to/tiny_bcl \
--csv=cellranger-tiny-bcl-simplesheet-1.2.0.csv如果使用第一种,则cellranger-tiny-bcl-simple-1.2.0.csv需要包含以下信息:
Lane,Sample,Index
1,test_sample,SI-P03-C9- Lane: 指定所在lane ID
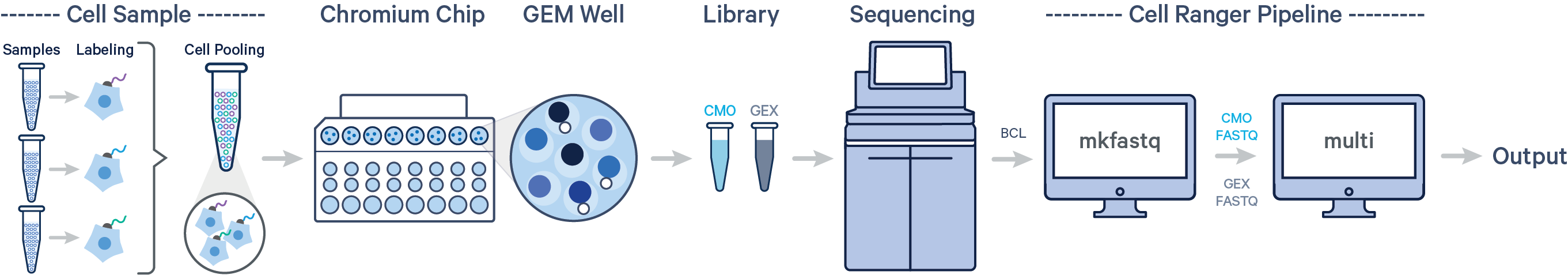
- Sample: 样本名称(来源于哪个GEM well,一般是一份生物样品,一个GEM well对应一个library,可以理解为下图芯片中的一个channel)
- Index: 指定index的名称,10X genomics的每个index代表4条具体的oligo序列
具体情况可以根据如下的测序芯片去理解。
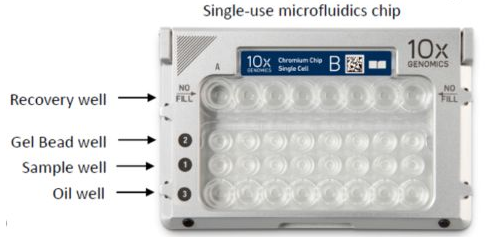
根据建库的不同可以分为如下操作:
- 两个不同的文库(或者说sample)在同一个flowcell上上机测序,执行一次
cellranger mkfastq,得到不同样本的数据
- 一个文库分两次或多次上机,需要根据上机的不同分别执行
cellranger mkfastq
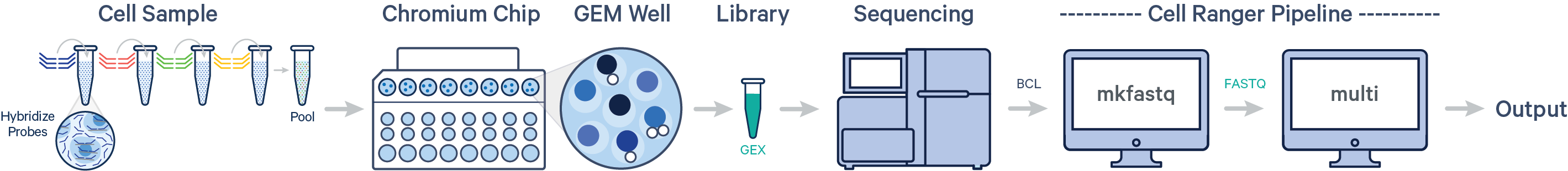
如果使用第二种方法,需要准备的信息就多一些(如下图所示),比如调整Reads中的序列长度,而使用简化版的csv文件,cell ranger可以识别所用试剂盒版本,然后自动化的调整reads长度。
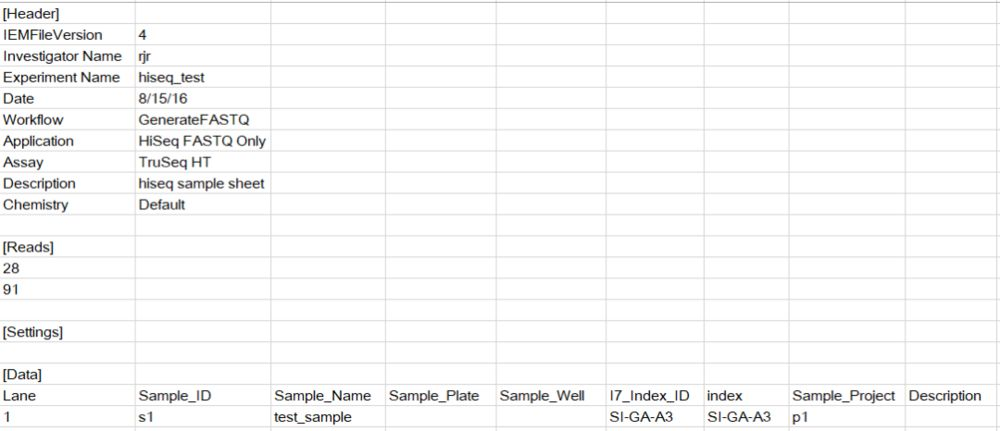
参数说明
--run 必选;BCL文件所在目录--id 可选;mkfastq将会生成相应目录,不接受绝对路径--samplesheet 可选. 样本名字、index等信息;--output-dir 指定FASTQ输出目录--localcores 运行的最大CPU数--localmem 运行的最大内存数
cellranger count
分析cellranger mkfastq或者其他方式得到fastq格式的文件。这个模块包括比对、过滤、barcoding计数以及UMI计数。可以生成barcode-UMI信息,进行聚类及基因表达分析。不过我们通常用到表达信息,使用其他方式进行聚类及其他单细胞相关分析。
在分析count之前需要先下载参考基因组
可以直接从10x Genmoics官网下载已经构建好的参考数据
curl -O "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz" # 人
curl -O "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCm39-2024-A.tar.gz" # 小鼠或使用cellranger mkref自己构建参考基因组
cellranger count --id=sample345 \
--transcriptome=/path/to/refdata-gex-GRCh38-2020-A \
--fastqs=/path/to/fastq \
--sample=mysample \
--localcores=8 \
--localmem=64输出结果包括以下信息:
Outputs:
- Run summary HTML: /outdir/outs/web_summary.html
- Run summary CSV: /opt/sample345/outs/metrics_summary.csv
- BAM: /opt/sample345/outs/possorted_genome_bam.bam
- BAM index: /opt/sample345/outs/possorted_genome_bam.bam.bai
- Filtered feature-barcode matrices MEX: /opt/sample345/outs/filtered_feature_bc_matrix
- Filtered feature-barcode matrices HDF5: /opt/sample345/outs/filtered_feature_bc_matrix.h5
......count分析完成之后可以打开web_summary.html文件查看报告,重点关注Summary,来判断文库构建及测序是否合格。
cellranger aggr
当实验中用到多个GEM well并且想要放在一起分析时需要先使用Cellranger count分析各自GEM well的fastq文件,然后再用Cellranger aggr将这些运行归一化到相同的测序深度,然后重新计算特征条形码矩阵并对组合数据进行分析。aggr管道可用于将来自多个样本的数据组合成一个实验范围内的特征条形码矩阵和分析。
相关信息
什么是GEM well
每个GEM well是10X芯片上的一个单独的区室,从barcode池(barcode whitelist)中随机获取barcode用于标记细胞。为了保证整合多个文库时barcode不发生冲突,通常会在barcode后面加一个数字,标记其来源的GEM well,如AGACCATTGAGACTTA-1和AGACCATTGAGACTTA-2,barcode序列一样,但来源于不同的GEM well,也是不同的细胞。
cellranger multi
cellranger multi用于分析Cell Multiplexing和Fixed RNA Profiling数据。它从cellranger mkfastq中获取FASTQ文件,并执行对齐、过滤、条形码计数和UMI计数。它使用Chromium细胞条形码生成特征条形码矩阵,确定簇,并执行基因表达分析。cellranger multi还支持特征条码数据的分析。
cellranger reanalyze
cellranger reanalyze采用由cellranger count、cellranger multi或cellrange aggr产生的特征条形码矩阵,并使用可调参数设置重新运行降维、聚类和基因表达算法。
使用说明
根据不同的情景,cellranger流程需要进行不同的调整,具体的分析操作可以分为以下几种:
- 单个样本,单次建库,单次上机
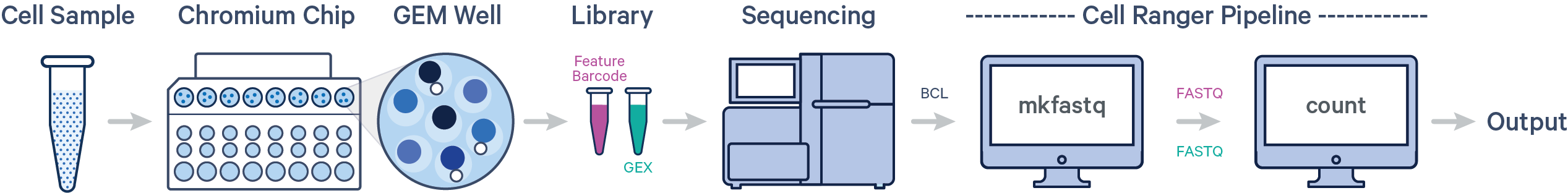
- 单个样本,单次建库,多次上机
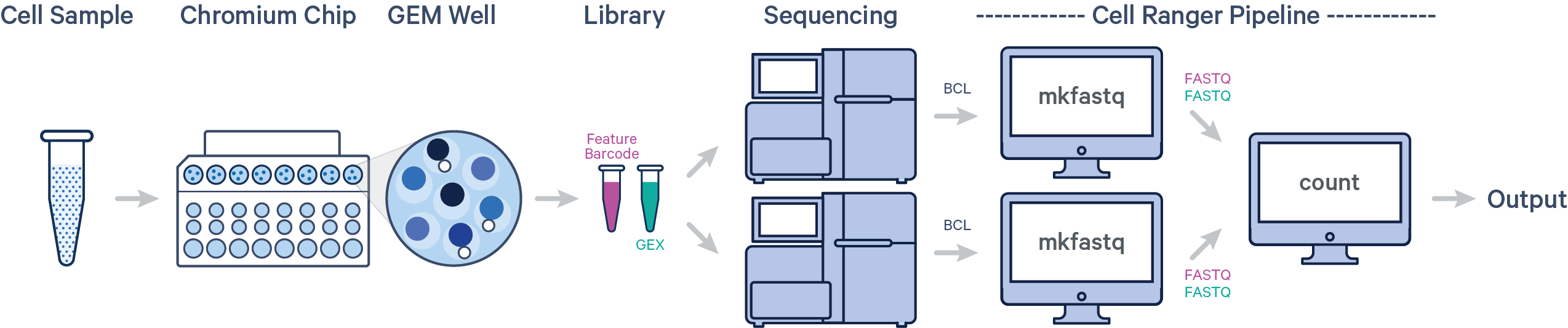
- 单个样本,多次建库,单次上机
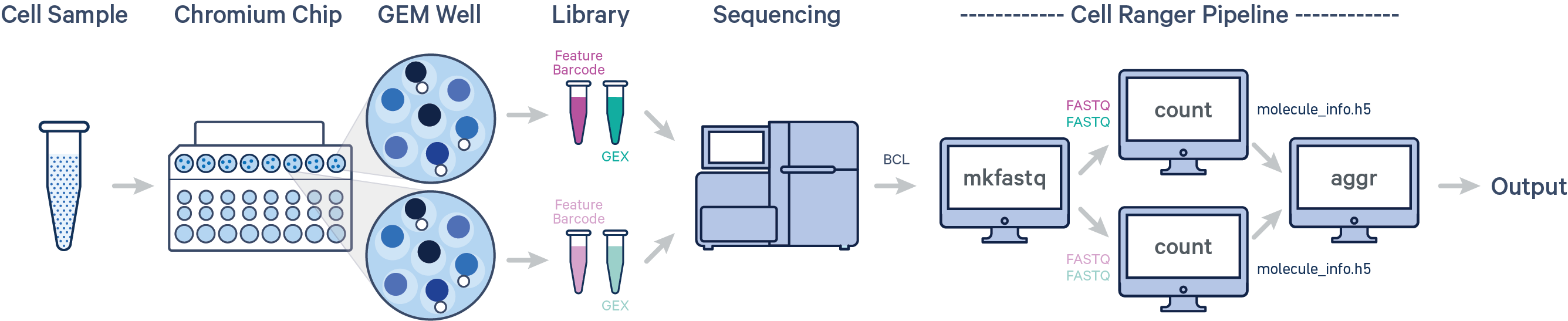
- 多个样本,多次建库,单次上机
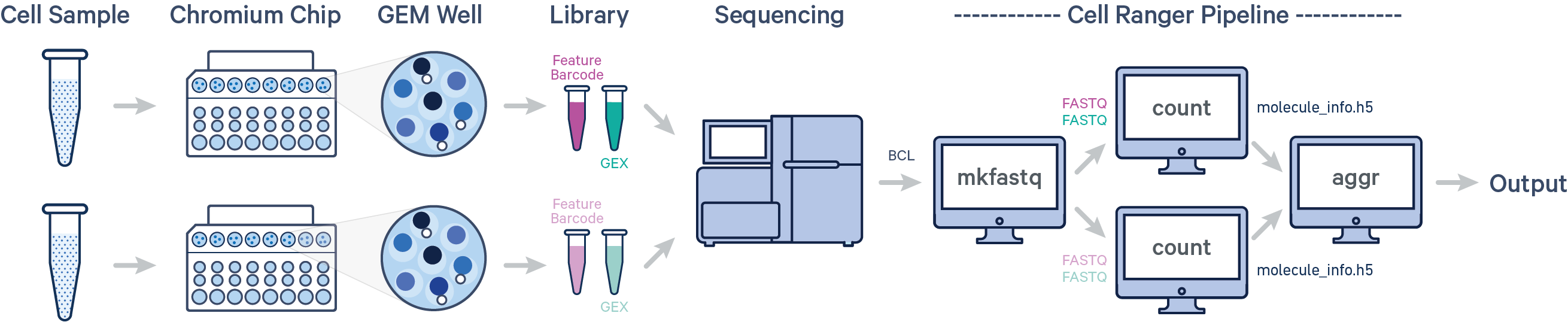
- 多个样本,单次建库,单次上机